Rainfall Plot
prettyRainfallPlot.RdPlot a rainfall plot for one sample. This function takes in the MAF data frame, or path to a custom MAF file. If non are specified, the SSM will be obtained through GAMBLR directly (with get_ssm_by_region).
prettyRainfallPlot(
this_sample_id,
label_ashm_genes = TRUE,
projection = "grch37",
chromosome,
this_maf,
maf_path,
zoom_in_region,
seq_type,
label_sv = FALSE
)Arguments
- this_sample_id
Sample id for the sample to display. This is argument is not required if you want a multi-sample plot but is otherwise needed.
- label_ashm_genes
Boolean argument indicating whether the aSHM regions will be labeled or not.
- projection
Specify projection (grch37 or hg38) of mutations. Default is grch37.
- chromosome
Provide one or more chromosomes to plot. The chr prefix can be inconsistent with projection and will be handled.
- this_maf
Specify custom MAF data frame of mutations.
- maf_path
Specify path to MAF file if it is not already loaded into data frame.
- zoom_in_region
Provide a specific region in the format "chromosome:start-end" to zoom in to a specific region.
- seq_type
Specify one of "genome" or "capture" when relying on the function to obtain mutations from a region (i.e. if you haven't provided a MAF or single sample_id)
- label_sv
Boolean argument to specify whether label SVs or not. Only supported if a specific chromosome or zoom in region are specified.
Value
a ggplot2 plot. Print it using print() or save it using ggsave()
Details
Create a sample-level rainfall plot visualizing single nucleotide substitutions mutations for selected chromosomes.
Examples
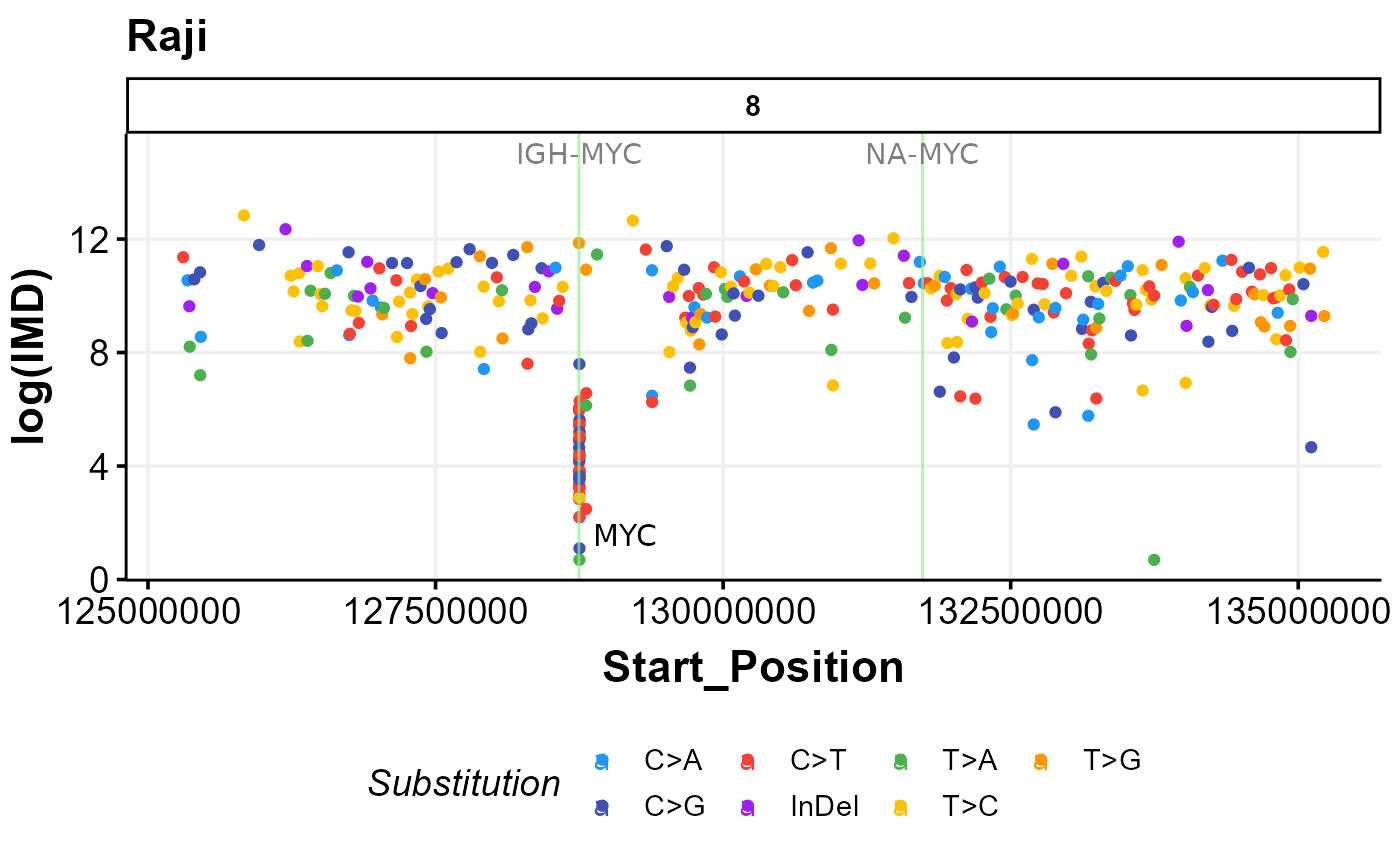
prettyRainfallPlot(this_sample_id = "Raji",
seq_type = "genome",
zoom_in_region = "8:125252796-135253201",
label_sv = TRUE)
#> MAF df or path to custom MAF file was not provided, getting SSM using GAMBLR ...
#> Warning: Column name 'GENE_PHENO' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'FILTER' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'flanking_bps' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'vcf_id' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'vcf_qual' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'gnomAD_AF' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'gnomAD_AFR_AF' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'gnomAD_AMR_AF' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'gnomAD_SAS_AF' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'vcf_pos' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'gnomADg_AF' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'blacklist_count' not found in column name header (case sensitive), skipping.
#> Warning: Column name 'FILTER' (colClasses[[1]][67]) not found
#> Warning: Column name 'flanking_bps' (colClasses[[1]][68]) not found
#> Warning: Column name 'vcf_id' (colClasses[[1]][69]) not found
#> Warning: Column name 'vcf_qual' (colClasses[[1]][70]) not found
#> Warning: Column name 'gnomADg_AF' (colClasses[[1]][71]) not found
#> Warning: Column name 'vcf_pos' (colClasses[[2]][11]) not found
#> Warning: Column name 'gnomAD_AF' (colClasses[[3]][20]) not found
#> Warning: Column name 'gnomAD_AFR_AF' (colClasses[[3]][21]) not found
#> Warning: Column name 'gnomAD_AMR_AF' (colClasses[[3]][22]) not found
#> Warning: Column name 'gnomAD_SAS_AF' (colClasses[[3]][28]) not found
#> Warning: Column name 'GENE_PHENO' (colClasses[[4]][5]) not found
#> Warning: Column name 'blacklist_count' (colClasses[[4]][6]) not found
#> Warning: Attempt to override column 92 <<MOTIF_SCORE_CHANGE>> of inherent type 'float64' down to 'bool8' ignored. Only overrides to a higher type are currently supported. If this was intended, please coerce to the lower type afterwards.
#> Getting combined manta + GRIDSS SVs using GAMBLR ...
#> Warning: There were 4 warnings in `dplyr::filter()`.
#> The first warning was:
#> ℹ In argument: `if (...) NULL`.
#> ℹ In group 1: `fusion = "IGH-MYC"`, `chromosomeN = chrom2`.
#> Caused by warning in `if (grepl("1", chromosomeN)) ...`:
#> ! the condition has length > 1 and only the first element will be used
#> ℹ Run `dplyr::last_dplyr_warnings()` to see the 3 remaining warnings.
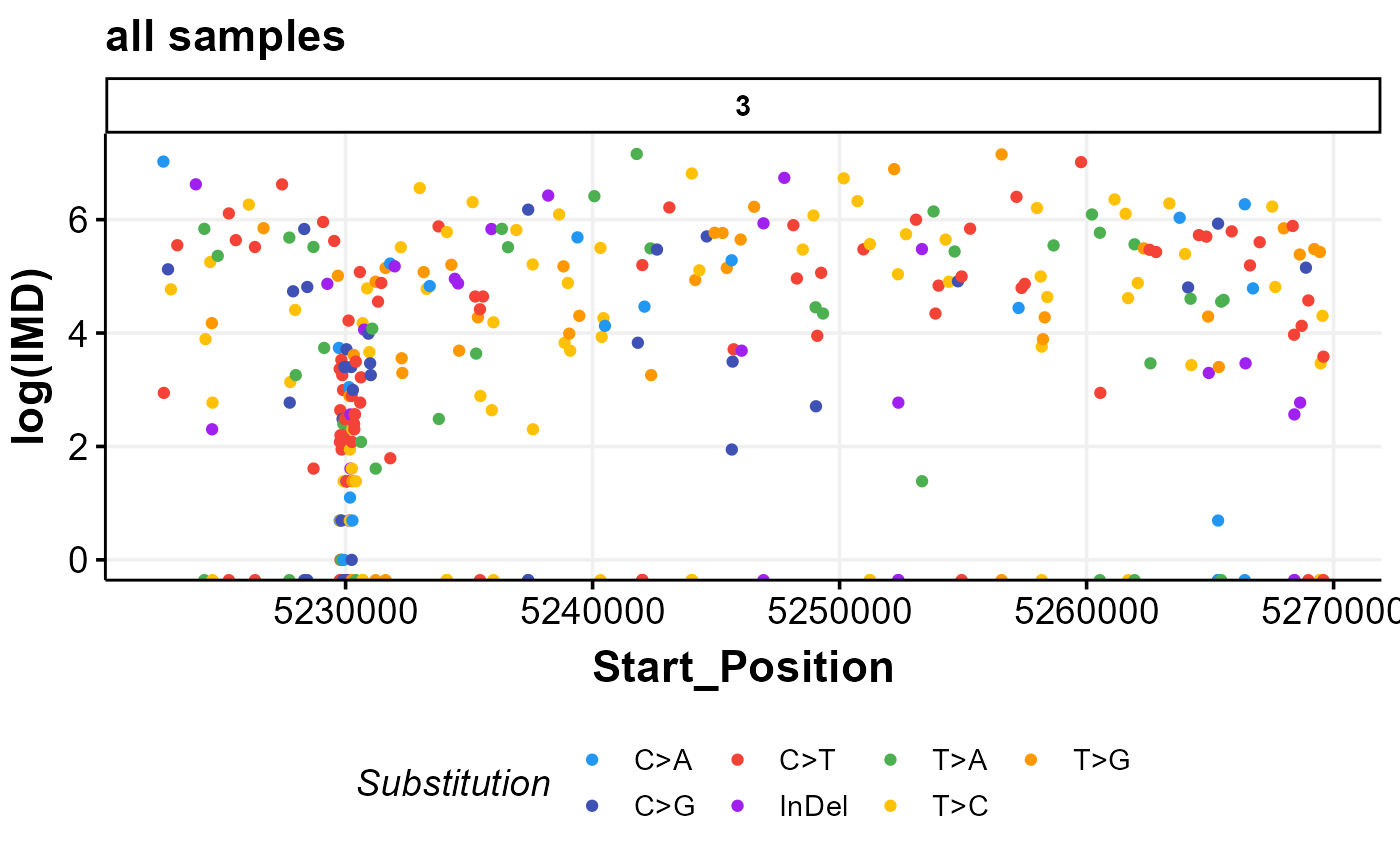
 #multi-sample rainfall plot for one gene region
prettyRainfallPlot(zoom_in_region = "chr3:5,221,286-5,269,723",
seq_type = "genome")
#> Warning: No sample_id was provided. Using all mutations in the MAF within your region!
#> Warning: after subsetting to a regions you requested to plot, there are no aSHM features to overlap on the final graph.
#> Will use all mutations for genome in this region: 3Will use all mutations for genome in this region: 5221286Will use all mutations for genome in this region: 5269723
#multi-sample rainfall plot for one gene region
prettyRainfallPlot(zoom_in_region = "chr3:5,221,286-5,269,723",
seq_type = "genome")
#> Warning: No sample_id was provided. Using all mutations in the MAF within your region!
#> Warning: after subsetting to a regions you requested to plot, there are no aSHM features to overlap on the final graph.
#> Will use all mutations for genome in this region: 3Will use all mutations for genome in this region: 5221286Will use all mutations for genome in this region: 5269723