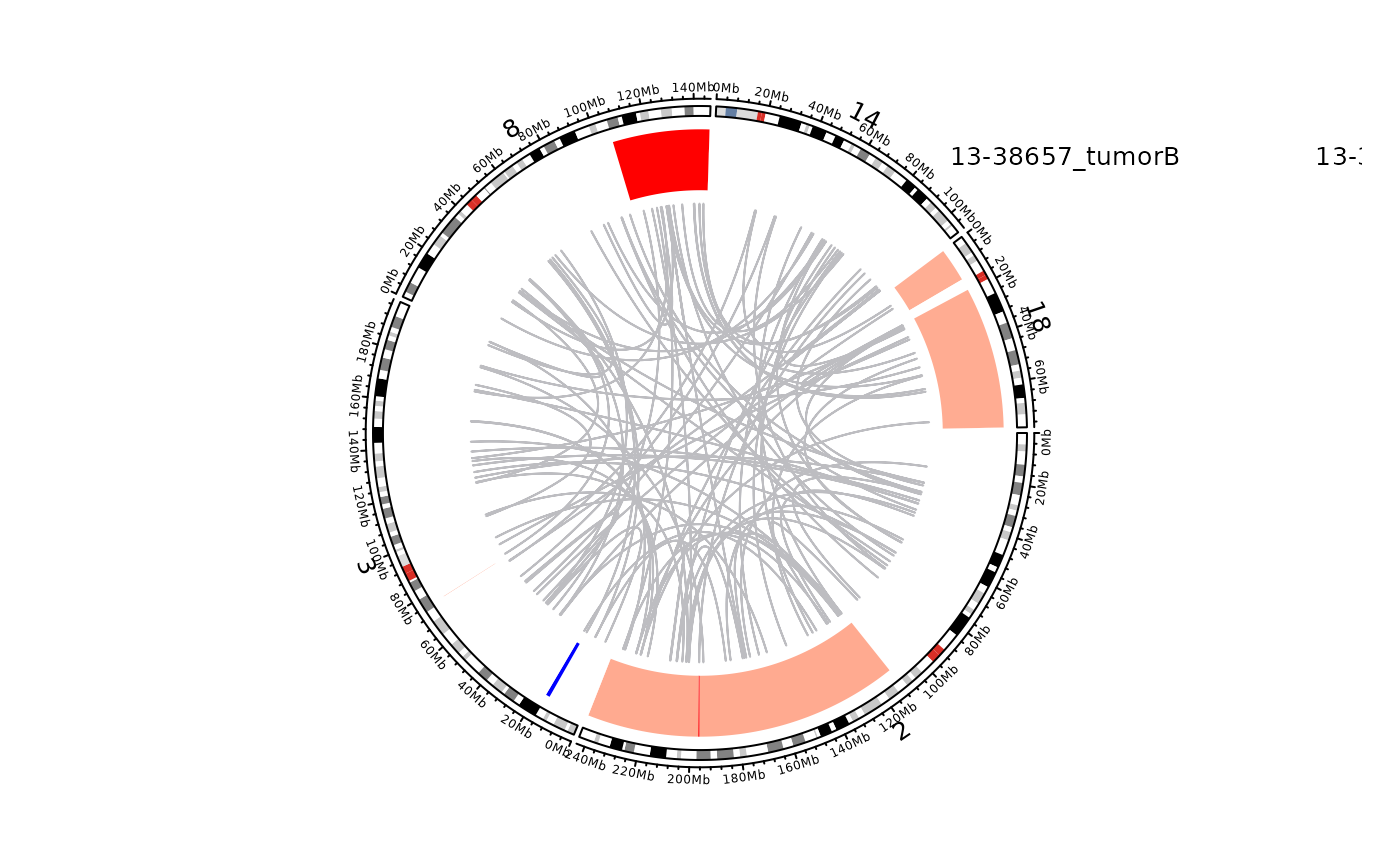
Sample-level Circos Plot
plot_sample_circos.RdPlot a sample-centric circos overview.
plot_sample_circos(
this_sample_id,
sv_df,
cnv_df,
ssm_df,
include_sv = TRUE,
include_ssm = FALSE,
legend_metadata_columns,
legend_metadata_names = c(),
include_cnv = TRUE,
this_projection = "grch37",
this_seq_type = "genome",
chrom_list,
label_genes,
auto_label_sv = FALSE
)Arguments
- this_sample_id
Sample ID for the sample to plot.
- sv_df
Optional data frame of SVs. If not provided this function will run
get_manta_svto retrieve SVs.- cnv_df
Optional data frame of CNVs. If not provided, this function will run
get_sample_cn_segmentsto retrieve CNVs.- ssm_df
This parameter does not do anything yet. Maybe it was meant to be implemented.
- include_sv
Default TRUE. (does not do anything yet).
- include_ssm
Defaul FALSE. (does not do anything yet).
- legend_metadata_columns
Column names from metadata
- legend_metadata_names
List of metadata names to be plotted.
- include_cnv
Default TRUE. (does not do anything yet).
- this_projection
The selected projection, default is grch37 and it's the only supported peojection.
- this_seq_type
Seq type for returned CN segments. One of "genome" (default) or "capture".
- chrom_list
List of chromosomes to be plotted. If not stated, chr1-22+X will bes used.
- label_genes
Gene labels (df, list or what type?)
- auto_label_sv
Default is FALSE
Value
Nothing
Details
This function takes a sample ID in the this_sample_id parameter.
Optionally, the user can supply already loaded data frames (SV, CNV, SSM) with the sv_df, cnv_df and ssm_df parameters.
Convenient Boolean parameteers are also avaialble for restricting the plot to specific mutation types (include_sv, include_cnv, and include_ssm).
Examples
plot_sample_circos(this_sample_id = "13-38657_tumorB",
legend_metadata_columns = c("pathology",
"lymphgen",
"COO_consensus",
"DHITsig_consensus",
"bcl2_ba",
"myc_ba"),
legend_metadata_names = c("pathology",
"LymphGen",
"COO",
"DHITsig",
"BCL2",
"MYC"),
chrom_list = c("chr2",
"chr3",
"chr8",
"chr14",
"chr18"))
#> WARNING! No SV calls found in flat-file for: 171116-PL02
#> WARNING! No SV calls found in flat-file for: 171447-PL01
#> WARNING! No SV calls found in flat-file for: 171451-PL01
#> [1] ">>>>>>>"
#> finding colour forDLBCL
#> [1] "<<<<<<<"
#> [1] "using alias to look up colours for pathology"
#> using pathology for pathology
#> adding:#479450
#> [1] ">>>>>>>"
#> finding colour forST2
#> [1] "<<<<<<<"
#> [1] "using alias to look up colours for lymphgen"
#> using lymphgen for lymphgen
#> adding:#C7371A
#> [1] ">>>>>>>"
#> finding colour forGCB
#> [1] "<<<<<<<"
#> [1] "using alias to look up colours for COO_consensus"
#> using coo for COO_consensus
#> adding:#F58F20
#> [1] ">>>>>>>"
#> finding colour forDHITsig-IND
#> [1] "<<<<<<<"
#> [1] "using alias to look up colours for DHITsig_consensus"
#> using coo for DHITsig_consensus
#> adding:#003049
#> [1] ">>>>>>>"
#> finding colour forPOS
#> [1] "<<<<<<<"
#> [1] "using alias to look up colours for bcl2_ba"
#> using pos_neg for bcl2_ba
#> adding:#c41230
#> [1] ">>>>>>>"
#> finding colour forPOS
#> [1] "<<<<<<<"
#> [1] "using alias to look up colours for myc_ba"
#> using pos_neg for myc_ba
#> adding:#c41230
#> DLBCL <NA> GCB DHITsig-IND POS POS
#> "#479450" NA "#F58F20" "#003049" "#c41230" "#c41230"
#> Error in gpar(fill = cols): could not find function "gpar"