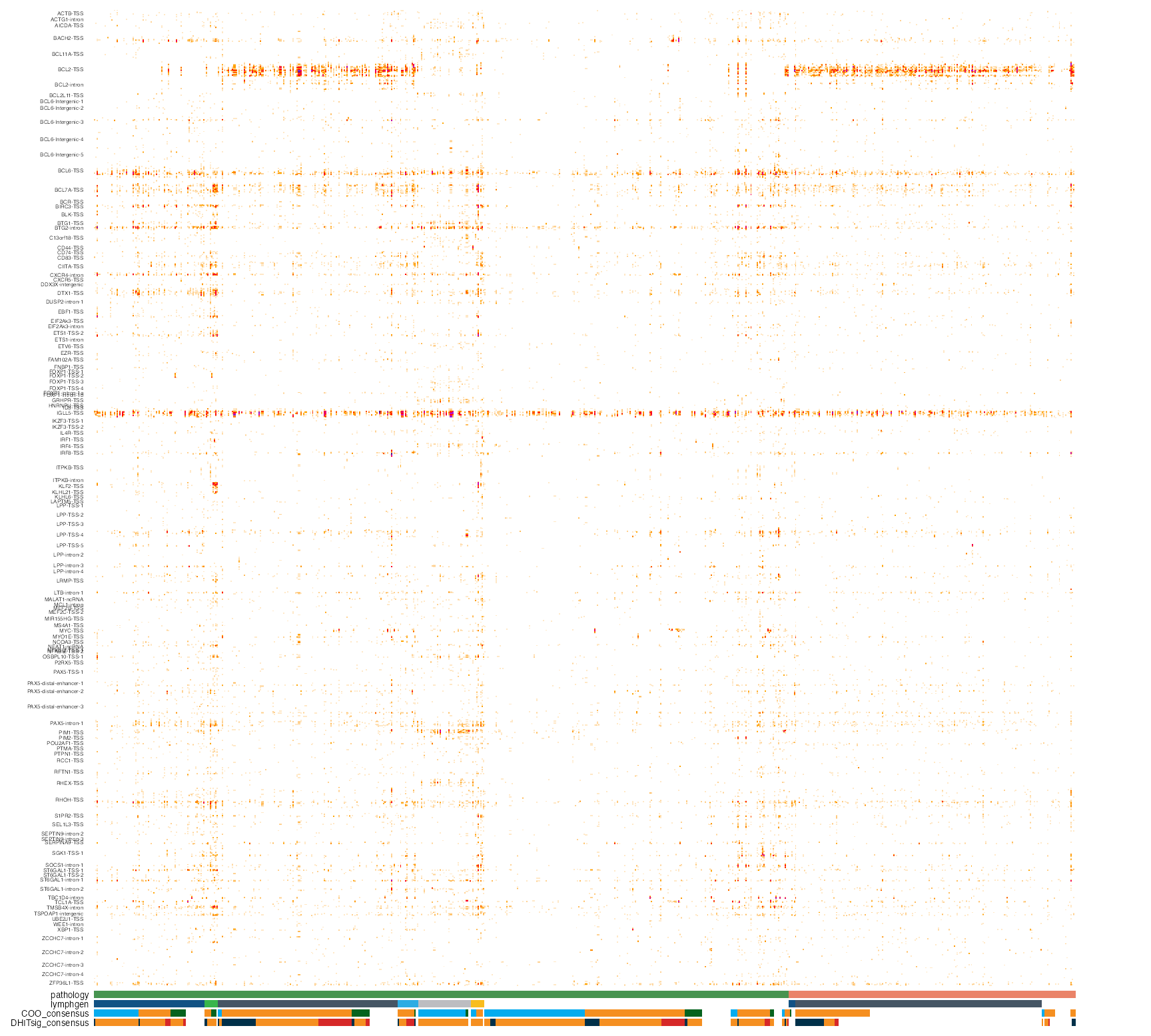
Pretty mutation density heatmap
prettyMutationDensity.RdObtain a heatmap of mutation counts across sliding windows for multiple regions.
Usage
prettyMutationDensity(
regions_list = NULL,
regions_bed = NULL,
these_samples_metadata = NULL,
these_sample_ids = NULL,
this_seq_type = c("genome", "capture"),
maf_data,
mut_freq_matrix,
projection,
region_padding = 1000,
drop_unmutated = FALSE,
metadataColumns = c("pathology"),
sortByMetadataColumns = NULL,
expressionColumns = NULL,
orientation = "sample_rows",
skip_regions,
only_regions,
customColours = NULL,
naColour = "white",
backgroundColour = "transparent",
slide_by = 100,
window_size = 500,
split_regions = TRUE,
min_count_per_bin = 0,
min_bin_recurrence = 5,
min_mut_tumour = 0,
region_fontsize = 8,
clustering_distance_samples = "euclidean",
cluster_samples = FALSE,
cluster_rows_heatmap,
split_samples_kmeans,
show_row_names = TRUE,
show_column_names = FALSE,
cluster_regions = FALSE,
show_gene_colours = FALSE,
label_regions_by = "name",
merge_genes = FALSE,
label_regions_rotate = 0,
legend_row = 3,
legend_col = 3,
show_legend = TRUE,
legend_direction = "horizontal",
legendFontSize = 10,
metadataBarHeight = 1.5,
metadataBarFontsize = 5,
metadataSide = "bottom",
region_annotation_name_side = "top",
sample_annotation_name_side = "left",
legend_side = "bottom",
returnEverything = FALSE,
from_indexed_flatfile = TRUE,
mode = "slms-3",
width,
height,
hide_annotation_name = FALSE,
use_raster = FALSE
)Arguments
- regions_list
Named vector of regions in the format c(name1 = "chr:start-end", name2 = "chr:start-end"). Only one (or none) between `regions_list` and `regions_bed` arguments should be provided. If neither regions_list nor regions_bed is specified, the function will use GAMBLR aSHM region information.
- regions_bed
Data frame of regions with four columns (chrom, start, end, name). Only one (or nome) between `regions_list` and `regions_bed` arguments should be provided.
- these_samples_metadata
Metadata with at least sample_id column. If not providing a maf data frame, seq_type is also required.
- these_sample_ids
Vector of sample IDs. Metadata will be subset to sample IDs present in this vector.
- this_seq_type
Optional vector of seq_types to include in heatmap. Default c("genome", "capture"). Uses default seq_type priority for samples with >1 seq_type.
- maf_data
Optional maf data frame. Will be subset to rows where Tumor_Sample_Barcode matches provided sample IDs or metadata table. If not provided, maf data will be obtained with get_ssm_by_regions().
- mut_freq_matrix
Optional matrix of binned mutation frequencies generated outside of this function, usually by [GAMBLR::calc_mutation_frequency_bin_regions].
- projection
Genome build the function will operate in. Ensure this matches your provided regions and maf data for correct chr prefix handling. Default grch37.
- region_padding
Amount to pad the start and end coordinates by. Default 1000
- drop_unmutated
Whether to drop bins with 0 mutations. If returning a matrix format, this will only drop bins with no mutations in any samples.
- metadataColumns
Mandatory character vector of metadata columns to use in heatmap annotation. Default c("pathology").
- sortByMetadataColumns
Optional character vector of metadata columns to order annotations by. Will be ordered by factor levels and sorted in the order specified. Default NULL.
- expressionColumns
Optional character vector of numeric metadata columns, usually gene expression, for heatmap annotation.
- orientation
Specify whether heatmap should have samples in rows ("sample_rows") or in columns ("sample_cols"). Default sample_rows.
- skip_regions
Optional character vector of genes to exclude from the default aSHM regions.
- only_regions
Optional character vector of genes to include from the default aSHM regions.
- customColours
Optional list of character vectors specifying colours for heatmap annotation with metadataColumns, e.g. list(pathology = c(DLBCL = "green", BL = "purple")). If left blank, the function will attempt to match heatmap annotations with existing colours from [GAMBLR::get_gambl_colours], or will default to the Blood colour palette.
- naColour
Colour to use for NA values in metadata/expression. Default "white".
- backgroundColour
Optionally specify the colour for heatmap bins with 0 mutations. Default grey90.
- slide_by
Slide size for sliding window. Default 100.
- window_size
Size of sliding window. Default 500.
- min_count_per_bin
Specify the minimum number of mutations per bin to be included in the heatmap. Only bins with all samples falling below this threshold will be dropped. Default 0.
- min_bin_recurrence
Specify how many samples a bin must be mutated in to be displayed. Default 5.
- min_mut_tumour
Specify how many bins a tumour must be mutated in to be displayed. Default 0.
- region_fontsize
Fontsize of region labels on the heatmap. Default 8.
- show_gene_colours
Boolean. Whether to add heatmap annotation colours for each region. Default FALSE.
- label_regions_by
Specify which feature of the regions to label the heatmap with. Heatmap will be split according to this value, and ordered by factor levels if the specified column is a factor. Default name.
- merge_genes
Set to TRUE to drop everything after "-" in the label to collpse regions from the same gene/locus. Default FALSE.
- label_regions_rotate
Specify degree by which the label in the previous parameter will be rotated. Default 0 (no rotation). The accepted values are 0, 90, 270.
- legend_row
Control aesthetics of the heatmap legend. Default 3.
- legend_col
Control aesthetics of the heatmap legend. Default 3.
- show_legend
Boolean. Default TRUE.
- legend_direction
Control aesthetics of the heatmap legend. Default "horizontal".
- legendFontSize
Control aesthetics of the heatmap legend. Default 10.
- metadataBarHeight
Optional argument to adjust the height of bar with annotations. The default is 1.5.
- metadataBarFontsize
Optional argument to control for the font size of metadata annotations. The default is 5.
- metadataSide
Default location for metadata is the bottom. Set to "top" if you want to move it
- legend_side
Control aesthetics of the heatmap legend. Default "bottom".
- returnEverything
Boolean. FALSE will plot the heatmap automatically. TRUE will return a heatmap object to allow further tweaking with the draw() function. Default FALSE.
- from_indexed_flatfile
Set to TRUE to avoid using the database and instead rely on flat files (only works for streamlined data, not full MAF details).
- mode
Only works with indexed flat files. Accepts 2 options of "slms-3" and "strelka2" to indicate which variant caller to use. Default is "slms-3".
- use_raster
Control whether ComplexHeatmap uses rastering. default: TRUE
Value
A table of mutation counts for sliding windows across one or more regions. May be long or wide.
Details
This function takes a metadata table with `these_samples_metadata` parameter and internally calls [GAMBLR::calc_mutation_frequency_bin_region] (that internally calls [GAMBLR::get_ssm_by_regions]). to retrieve mutation counts for sliding windows across one or more regions and generate a heatmap. May optionally provide any combination of a maf data frame, existing metadata, or a regions data frame or named vector.
Examples
library(GAMBLR.open)
# get meta data
my_meta <- get_gambl_metadata() %>%
dplyr::filter(pathology %in% c("FL","DLBCL"), seq_type != "mrna") %>%
check_and_clean_metadata(duplicate_action = "keep_first")
#> Using the bundled metadata in GAMBLR.data...
#> Duplicate rows (keeping first occurrence) for 'sample_id' and 'seq_type' have been dropped.
# get ashm regions of a set of genes.
my_regions = create_bed_data(GAMBLR.data::grch37_ashm_regions,
fix_names = "concat",
concat_cols = c("gene","region"),sep = "-")
# create heatmap of mutation counts for the specified regions
meta_columns <- c("pathology",
"lymphgen",
"COO_consensus",
"DHITsig_consensus")
suppressMessages(
suppressWarnings({
prettyMutationDensity(
regions_bed = my_regions,
these_samples_metadata = my_meta,
metadataColumns = meta_columns,
orientation="sample_columns",
sortByMetadataColumns = meta_columns,
projection = "grch37",
backgroundColour = "transparent",
show_legend = FALSE,
region_fontsize = 3)
}))