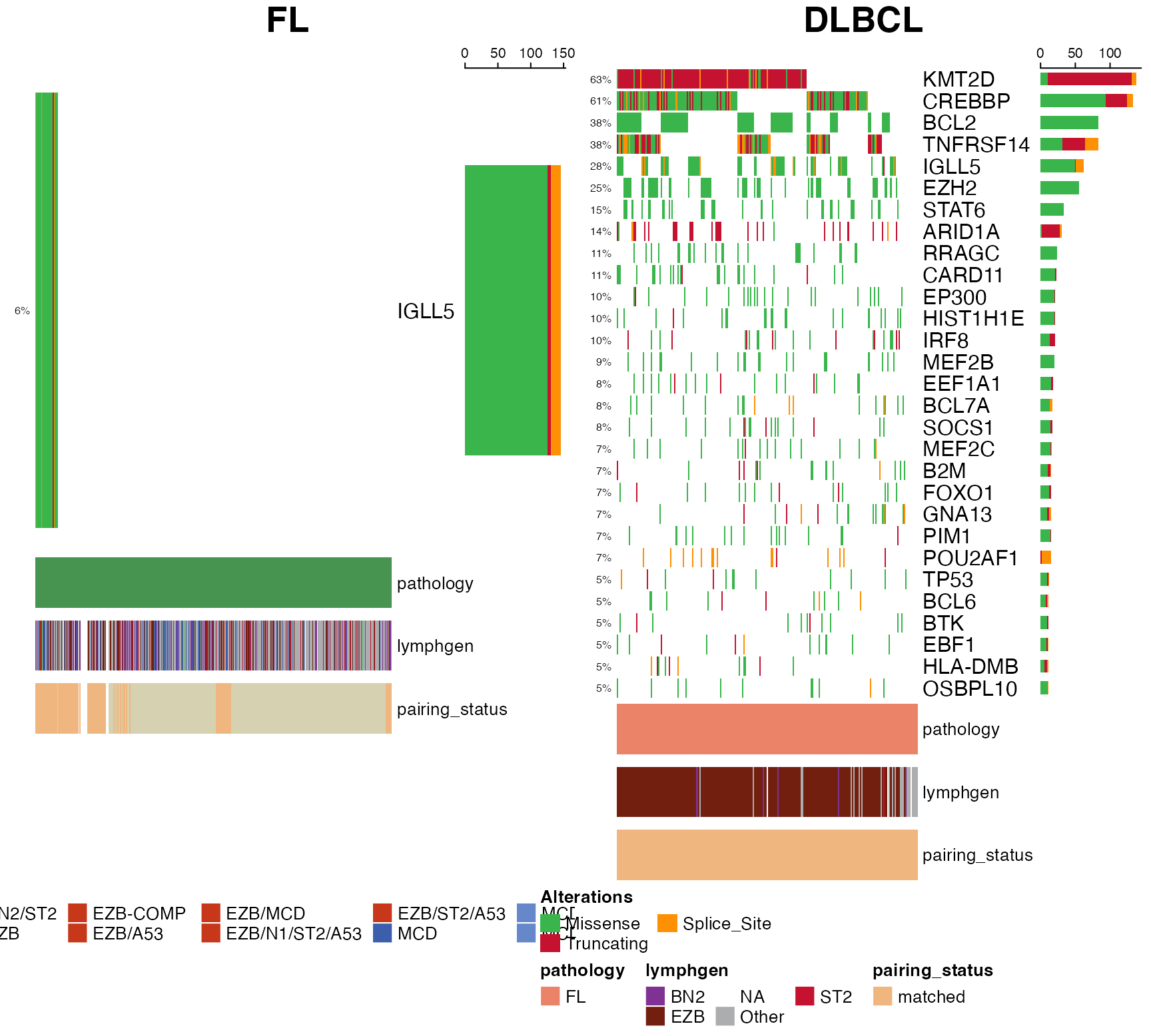
Side-by-side Oncoplots
prettyCoOncoplot.Rd`prettyCoOncoplot` returns ggplot-compatible figure of 2 [GAMBLR.viz::prettyOncoplot] side-by-side.
Arguments
- maf
Required argument. A data frame containing the mutations you want to plot on both oncoplots.
- metadata
Required argument. A data.frame with metadata for both oncoplots.
- comparison_column
Required: the name of the metadata column containing the comparison values.
- comparison_values
Optional: If the comparison column contains more than two values or is not a factor, specify a character vector of length two in the order you would like the factor levels to be set, reference group first.
- label1
Optional argument. Label to be shown as a title for the oncoplot #1.
- label2
Optional argument. Label to be shown as a title for the oncoplot #2.
- ...
`prettyOncoplot` arguments, see that function for more info on avaialble parameters.
Details
This function will generate a graphic displaying 2 oncoplots side-by-side. Optionally user can annotate each oncoplot with it's own title that will be displayed at the top. All the arguments recognized by [GAMBLR.viz::prettyOncoplot] are supported and can be specified when calling this function. For both oncoplots the same specified parameters will be applied (e.g. genes to display, split columns, font size, top annotation etc). If the provided argument is not recognized by [GAMBLR.viz::prettyOncoplot], it will be discarded. If you want a specific order of oncoplots on the left and right, please ensure the argument `comparison_column` is a factor with first level being the group you want to be plotted on the left side. For developers: new arguments added to [GAMBLR.viz::prettyOncoplot] in the future are expected to be out-of-the-box compatible with this function nd would not need code modifications.
Examples
library(GAMBLR.open)
#get data for plotting
meta <- get_gambl_metadata() %>%
GAMBLR.helpers::check_and_clean_metadata(duplicate_action = "keep_first")
#> Using the bundled metadata in GAMBLR.data...
#> Duplicate rows (keeping first occurrence) for 'sample_id' and 'seq_type' have been dropped.
meta <- meta %>%
dplyr::filter(
pathology %in% c("DLBCL", "FL")
)
ssm <- get_coding_ssm(
these_samples_metadata = meta
)
#> Using the bundled SSM calls (.maf) calls in GAMBLR.data...
#> id_ease: WARNING! 1783 samples in the provided metadata were removed because their seq types are not the same as in the `set_type` argument. Use `verbose = TRUE` to see their IDs.
#> after linking with metadata, we have mutations from 546 samples
suppressMessages(
suppressWarnings({
#build plot
prettyCoOncoplot(
maf = ssm,
metadata = meta,
comparison_column = "pathology",
comparison_values = c("DLBCL","FL"),
genes=dplyr::filter(lymphoma_genes,
FL_Tier==1 | DLBCL_Tier==1) %>%
dplyr::pull(Gene),
metadataColumns = c(
"pathology",
"lymphgen",
"pairing_status"
),
metadataBarHeight = 10,
fontSizeGene = 12,
metadataBarFontsize = 10,
legend_row = 2,
label1 = "FL",
label2 = "DLBCL",
simplify_annotation =TRUE,
minMutationPercent = 5
)
}))